Implementing SAKURA
In this tutorial, we go through the basic steps for implementing SAKURA, using a dataset of ~5k Peripheral Blood Mononuclear Cells (PBMC) freely available from 10XGenomics with marker genes CD8A and CD8B as the knowledge input to guide dimensionality reduction. The processed data can be found here in the documentation repository.
Inputs of SAKURA
Data preprocessing
We first include a data preprocessing pipeline using R and the Seurat package, which starts by reading in the data. The Read10X_h5() function in Seurat reads in the HDF5 file that contains single-cell RNA sequencing (scRNA-seq) data. We use the count matrix to initialize a Seurat object with quality control parameters:
library(Seurat)
library(Matrix)
library(tidyverse)
library(ggplot2)
data <- Read10X_h5(filename = "./tests/pbmc5k/raw/5k_Human_Donor1_PBMC_3p_gem-x_5k_Human_Donor1_PBMC_3p_gem-x_count_sample_filtered_feature_bc_matrix.h5")
# Retains genes detected in at least 3 cells, cells with at least 200 detected genes
seurat_object <- CreateSeuratObject(counts=data, project="pbmc5k", min.cells=3, min.features=200)
Cell Type Filtering
Next, we read in the cell type information and filter a few cell types in the PBMC_5k dataset for simplicity, and then integrate corresponding cell type information into Seurat object metadata.
cell_type <- read.csv("./tests/pbmc5k/raw/cell_types.csv",row.names = 1,header=TRUE)
cur_cell <- rownames(seurat_object@meta.data)
cur_celltype <- cell_type[cur_cell, ]
seurat_object@meta.data <- cbind(seurat_object@meta.data, cur_celltype)
cell_types_to_remove <- c("glial cell", "hematopoietic cell", "hematopoietic precursor cell", "mast cell", "stem cell")
cells_to_keep <- !seurat_object@meta.data$coarse_cell_type %in% cell_types_to_remove
seurat_filtered <- seurat_object[, cells_to_keep]
Note
From 10XGenomics, cell type annotation (https://www.10xgenomics.com/support/software/cell-ranger/latest/algorithms-overview/cr-cell-annotation-algorithm) is currently in beta and relies on the Chan Zuckerberg CELL by GENE reference. As this reference is community-driven it may not cover all tissue types, which could impact your results.
Normalization and Feature Selection
We then perform minimal pre-processing of the data, including log-transformation of the raw UMI counts using NormalizeData(), selecting the 10,000 genes with highest variance using FindVariableFeatures(), and scaling the data based on the high variance genes using ScaleData().
seurat_filtered <- seurat_filtered %>% NormalizeData() %>% FindVariableFeatures(nfeatures = 10000)
seurat_filtered <- seurat_filtered %>% ScaleData(features = VariableFeatures(seurat_filtered))
seurat.hv10k <- seurat_filtered[VariableFeatures(seurat_filtered),]
Export Preprocessed Data
With 10k highly variable genes subset, we export key data components as input data of SAKURA:
genenames_hv10k.csv: List of the 10k highly variable gene names
cell_names.csv: Filtered cell barcodes
pheno_df.csv: Cell metadata including cell type annotations
lognorm_hv10k.mtx: Normalized expression matrix in Matrix Market format
write.csv(rownames(seurat.hv10k),"./tests/pbmc5k/processed/genenames_hv10k.csv")
write.csv(colnames(seurat.hv10k),"./tests/pbmc5k/processed/cell_names.csv")
write.csv(seurat.hv10k@meta.data,"./tests/pbmc5k/processed/pheno_df.csv")
writeMM(seurat.hv10k@assays$RNA$data,"./tests/pbmc5k/processed/lognorm_hv10k.mtx")
Users can prepare the pre-processed example dataset as input for SAKURA or use their own datasets. Training and testing with demo configuration files on this dataset will cost less than 1 hour on the Intel Xeon CPU E5-2630 v2 @ 2.60GHz, which can be substantially improved with GPUs.
Running SAKURA with the example dataset
SAKURA uses comprehensive JSON configuration files to control all aspects of the training process. This section demonstrates how to run SAKURA using the provided example dataset PBMC5k and configuration files.
./tests/pbmc5k/config.json
./tests/pbmc5k/signature_config.json
Since we have installed SAKURA in installation, we can now run SAKURA as a python module:
(sakura) ~/.../SAKURA/$ python -m sakura -c ./test/pbmc5k/config.json -v True &> ./test/pbmc5k/console.log
Command Breakdown:
-c ./test/pbmc5k/config.json: Specifies the path to configuration file
-v True: Enables verbose logging for detailed progress information
&> ./test/pbmc5k/console.log: Redirects both standard output and error to log file
Note
See sakura.sakuraAE.sakuraAE.train(), sakura.sakuraAE.sakuraAE.train_hybrid_fastload(),
sakura.sakuraAE.sakuraAE.train_story() for more details of different training <action>s.
Below we break down some of the key parameters in each section of the example configuration files:
Basic Configuration
{
"remarks": "",
"log_path": "./test/pbmc5k/log/",
"reproducible": "True",
"rnd_seed": 3407,
Parameters:
remarks: User comments or notes about this configuration
log_path: Directory path for storing training logs and outputs
reproducible: When set to “True”, ensures reproducible results using the specified random seed
rnd_seed: Random seed (3407) for reproducible random number generation
Dataset Configuration
Related API: sakura.dataset
"dataset": {
"type": "rna_count_sparse",
"gene_expr_MM_path": "./tests/pbmc5k/processed/lognorm_hv10k.mtx",
"gene_name_csv_path": "./tests/pbmc5k/processed/genenames_hv10k.csv",
"cell_name_csv_path": "./tests/pbmc5k/processed/cell_names.csv",
"pheno_csv_path": "./tests/pbmc5k/processed/pheno_df.csv",
"pheno_df_dtype": {
"Batch": "string",
"Main_cluster_name": "string"
},
"pheno_df_na_filter": "False",
"expr_mat_pre_slice": "False",
"signature_config_path": "./tests/pbmc5k/processed/signature_config.json",
"selected_signature": ["cd8"]
},
Parameters:
type: Data format type (“rna_count_sparse” for PBMC5k sparse RNA count matrices)
gene_expr_MM_path: Path to normalized gene expression matrix in Matrix Market format
gene_name_csv_path: Path to CSV file containing gene names
cell_name_csv_path: Path to CSV file containing cell barcodes/identifiers
pheno_csv_path: Path to CSV file containing cell phenotype metadata
pheno_df_dtype: Data types for phenotype columns (Batch and cell type annotations as strings)
pheno_df_na_filter: Whether to filter out NA values in phenotype data
expr_mat_pre_slice: Whether the expression matrix is pre-sliced
signature_config_path: Path to signature configuration file defining signature learning task
selected_signature: Name list of signatures to use (here, we use ‘CD8A’, ‘CD8B’ and name them ‘cd8’)
Note
Similarly, users can include optional phenotype learning task configuration JSON file with
pheno_meta_path and selected_pheno. See signature_config for more details.
Hardware and Data Splitting
Related API: sakura.sakuraAE.sakuraAE.generate_splits() and sakura.utils.data_splitter.DataSplitter
"device": "cpu",
"overall_train_test_split": {
"type": "auto",
"train_dec": 5,
"seed": 3407
},
Parameters:
device: Computation device (“cpu” for CPU-only, “cuda” for GPU acceleration)
overall_train_test_split: Configuration for splitting data into training and testing sets
type: “auto” for automatic splitting
train_dec: Training set ratio denominator (5 = 80% training, 20% testing)
seed: Random seed for reproducible data splitting
Model Architecture
Related API: structure settings - sakura.models.extractor.Extractor
and loss/regularization settings - sakura.model_controllers.extractor_controller.ExtractorController
"main_latent": {
"encoder_neurons": 200,
"decoder_neurons": 200,
"latent_dim": 50,
"loss": {
"L2": {
"type": "MSE",
"init_weight": 1.0,
"progressive_const": 1.0,
"progressive_start_epoch": 100
},
"L1": {
"type": "L1",
"init_weight": 1.0,
"progressive_const": 1.0,
"progressive_start_epoch": 100
}
},
"regularization": {
"uniform_shape_unsupervised": {
"type": "SW2_uniform",
"init_weight": 0.0001,
"progressive_const": 1.01,
"progressive_start_epoch": 1,
"max_weight": 1.0,
"SW2_num_projections": 50,
"uniform_low": -10,
"uniform_high": 10
}
}
},
Parameters:
encoder_neurons: Number of neurons in encoder hidden layers (200)
decoder_neurons: Number of neurons in decoder hidden layers (200)
latent_dim: Dimensionality of the latent space (50)
loss: Reconstruction loss configuration
L2: Mean Squared Error loss with progressive weighting
L1: L1 loss for robust reconstruction
regularization: Regularization terms to shape the latent space
uniform_shape_unsupervised: Sliced Wasserstein distance regularization to enforce uniform distribution
Optimizer Configuration
Related API: sakura.model_controllers.extractor_controller.ExtractorController.setup_optimize()
"optimizer": {
"type": "RMSProp",
"RMSProp_lr": 0.001,
"RMSProp_alpha": 0.9
},
Parameters:
type: Optimization algorithm (“RMSProp”)
RMSProp_lr: Learning rate (0.001)
RMSProp_alpha: Smoothing constant for RMSProp (0.9)
Training Pipeline (Story)
Related API: sakura.sakuraAE.sakuraAE.train_hybrid()
The story section defines the complete training workflow:
"story": [
{
"action": "train_hybrid",
"ticks": 5000,
"hybrid_mode": "interleave",
"split_configs": {
"main_lat_reconstruct": {
"use_split": "overall_train",
"batch_size": 100,
"train_main_latent": "True",
"train_pheno": "False",
"train_signature": "False"
},
"cd8_focused": {
"use_split": "overall_train",
"batch_size": 100,
"train_main_latent": "False",
"train_pheno": "False",
"train_signature": "True",
"selected_signature": {
"cd8": {
"loss": "*",
"regularization": "*"
}
}
}
},
"prog_loss_weight_mode": "epoch_end",
Training Splits Strategy:
action: Training mode (“train_hybrid” for both reconstruction and signature regression training)
ticks: Total number of training ticks (batches), 5000 is set considering the size of pbmc5k
hybrid_mode: “interleave” alternates between different training tasks
main_lat_reconstruct: Main autoencoder reconstruction training
batch_size: 100 cells per batch
cd8_focused: Signature-guided training, use cd8 related signature to guide latent space organization and applies losses and regularizations according to signature_config.
prog_loss_weight_mode: “epoch_end” controls loss weight updated at the end of each epoch
Note
See also sakura.sakuraAE.sakuraAE.train(), sakura.sakuraAE.sakuraAE.train_hybrid_fastload(),
sakura.sakuraAE.sakuraAE.train_story() for more details of different training <action>s.
Logging and Checkpointing
Related API: sakura.utils.logger.Logger and sakura.sakuraAE.sakuraAE.save_checkpoint()
Save model checkpoints every 500 ticks:
"make_logs": "True",
"log_prefix": "hybrid",
"save_raw_loss": "True",
"log_loss_groups": ["loss", "regularization", "loss_raw", "regularization_raw"],
"perform_checkpoint": "True",
"checkpoint_on_segment": "True",
"checkpoint_segment": 500,
"checkpoint_prefix": "checkpoint_",
"checkpoint_save_arch": "True",
"checkpoint_every_epoch": "False",
Testing and Latent dumping
Related API: sakura.sakuraAE.sakuraAE.save_checkpoint() and sakura.utils.logger.Logger
Perform tests on different data splits and save latent representations according to test configurations every 500 ticks:
"perform_test": "True",
"test_segment": 500,
"tests": [
{
"on_split": "all",
"make_logs": "False",
"dump_latent": "True",
"latent_prefix": "all_cell_all_latent"
},
{
"on_split": "overall_test",
"make_logs": "True",
"log_prefix": "overall_test",
"dump_latent": "True",
"latent_prefix": "overall_test_all_latent"
},
{
"on_split": "overall_train",
"make_logs": "False",
"dump_latent": "True",
"latent_prefix": "overall_train_all_latent"
}
]
Signature Configuration
In this tutorial, we use marker gene expression signature to incorporate biological prior knowledge into SAKURA training process.
The ./pbmc5k/signature_config.json file defines one biological signature that SAKURA should respect during training.
Example: CD8 T-cell Signature
{
"cd8": {
"remarks": "major CD8 T cells marker(s)",
"signature_list": [
"CD8A",
"CD8B"
],
Signature Definition:
cd8: Signature identifier used throughout the configurations
signature_list: Array of gene names (CD8A, CD8B) that define this signature
Note
Genes of signatures should be contained in the input data.
Signature Processing Configuration
Related API: sakura.sakuraAE.sakuraAE.setup_dataset() and sakura.models.extractor.Extractor
"exclude_from_input": "False",
"signature_lat_dim": 50,
"signature_out_dim": 2,
"pre_procedure": [],
"post_procedure": [
{
"type": "ToTensor"
}
],
Processing Parameters:
exclude_from_input: Whether to remove signature genes from input data
"False": Signature genes remain in the main expression matrix
"True": Signature genes are excluded to prevent data leakage
signature_lat_dim: Dimensionality of signature-specific latent space (50)
signature_out_dim: Output dimension for signature prediction (2), shouldcorresponds to the number of genes in
signature_list
Signature Splitting
Related API: sakura.sakuraAE.sakuraAE.generate_splits() and sakura.utils.data_splitter.DataSplitter
"split": {
"type": "none"
},
- Split Configuration:
type:"none"- No special splitting for this signature
Note
See also data_splitting for similar format.
Signature Model Architecture
Related API: structure settings - sakura.models.extractor.Extractor
and loss/regularization settings - sakura.model_controllers.extractor_controller.ExtractorController
"model": {
"type": "FCRegressor",
"hidden_neurons": 5,
"attach": "True",
"attach_to": "main_lat"
},
"loss": {
"regression_MSE": {
"type": "MSE",
"progressive_mode": "increment",
"progressive_const": 0.01,
"progressive_start_epoch": 50,
"init_weight": 0.0,
"max_weight": 1.0
},
"regression_L1": {
"type": "L1",
"progressive_mode": "increment",
"progressive_const": 0.01,
"progressive_start_epoch": 50,
"init_weight": 0.0,
"max_weight": 1.0
},
"regression_cosine": {
"type": "Cosine",
"progressive_mode": "increment",
"progressive_const": 0.01,
"progressive_start_epoch": 50,
"init_weight": 0.0,
"max_weight": 1.0
}
},
"regularization": {
}
Signature Model Configuration:
type:"FCRegressor"- Fully Connected Regression model; Alternative:"FCClassifier"for classification tasks
hidden_neurons: 5 - Number of neurons in hidden layer
attach:"True"- Connect this model to the main network
attach_to:"main_lat"- Connect to the main latent space, allowing signature model to influence main representation learning
Outputs of SAKURA
In this section, we simply explain the outputs generated by SAKURA and show how to retrieve them for downstram analysis.
Configuration Archives
Following configuration, SAKURA saves settings and metadata to the log folder for reproducibility:
signature_config.json: A complete copy of the signature configuration
gene_meta.json: Metadata about genes used in the analysis
pheno_config.json: Phenotype data configuration (empty file here)
splits.pkl: Serialized Python object containing the exact train/test split indices used during SAKURA training
Checkpoint and TensorBoard Event Files
Following configuration, SAKURA saves model checkpoints at regular intervals during training and records hierarchical losses to TensorBoard:
Related API: sakura.sakuraAE.sakuraAE.save_checkpoint() and sakura.utils.logger.Logger
checkpoint_tick_[500~5000].pth: Checkpoint every 500 ticks with complete training state, including model weights, optimizer state, and training metadata
events.out.tfevents.*: TensorBoard compatible event files containing training metrics, loss curves, and histograms
Cell Embedding Outputs
Following the latent_dumping configuration, SAKURA generates cell embedding outputs including both main reconstruction latents and signature latents every 5000 tickss during testing on different splits:
[#epoch]_all_cell_all_latent.csv: 50d Main and 50d signature latent space coordinates for all cells in the dataset
[#epoch]_overall_test_all_latent.csv: 50d Main and 50d signature latent space coordinates for all cells in the testing set``[#epoch]_overall_train_all_latent.csv`: 50d Main and 50d signature latent space coordinates for all cells in the training set
Since the signature latent space is attached to the main latent space as configured, both components share the same 50d coordinate outputs.
Therefore, we use the [last training epoch]_all_cell_all_latent.csv and take the first 50 dimensions as the final cell embedding for downstream analysis.
Comparative Analysis: SAKURA vs 10x Genomics Cell Ranger
This final section demonstrates how to simply compare SAKURA’s final cell embeddings with 10x Genomics Cell Ranger analysis results using Seurat.
Loads 10xGenomics Cell Ranger Analysis
We first load the pre-computed UMAP coordinates and cluster assignments from Cell Ranger and show comparative visualizations (UMAP) colored by Cell Ranger coarse cell type annotations and the cluster assignments.
umap <- read.csv("./tests/pbmc5k/projection.csv",row.names = 1,header=TRUE)
cluster <- read.csv("./tests/pbmc5k/clusters.csv",row.names = 1,header=TRUE)
cur_umap <- umap[cur_cell, ]
cur_cluster <- cluster[cur_cell, ]
umap_filtered <- cur_umap[cells_to_keep,]
cluster_filetered <- cur_cluster[cells_to_keep]
seurat.hv10k[["crumap"]] <- CreateDimReducObject(embeddings = umap_filtered %>% as.matrix, key = "crumap_", assay = DefaultAssay(seurat.hv10k))
Idents(seurat.hv10k) <- cluster_filetered
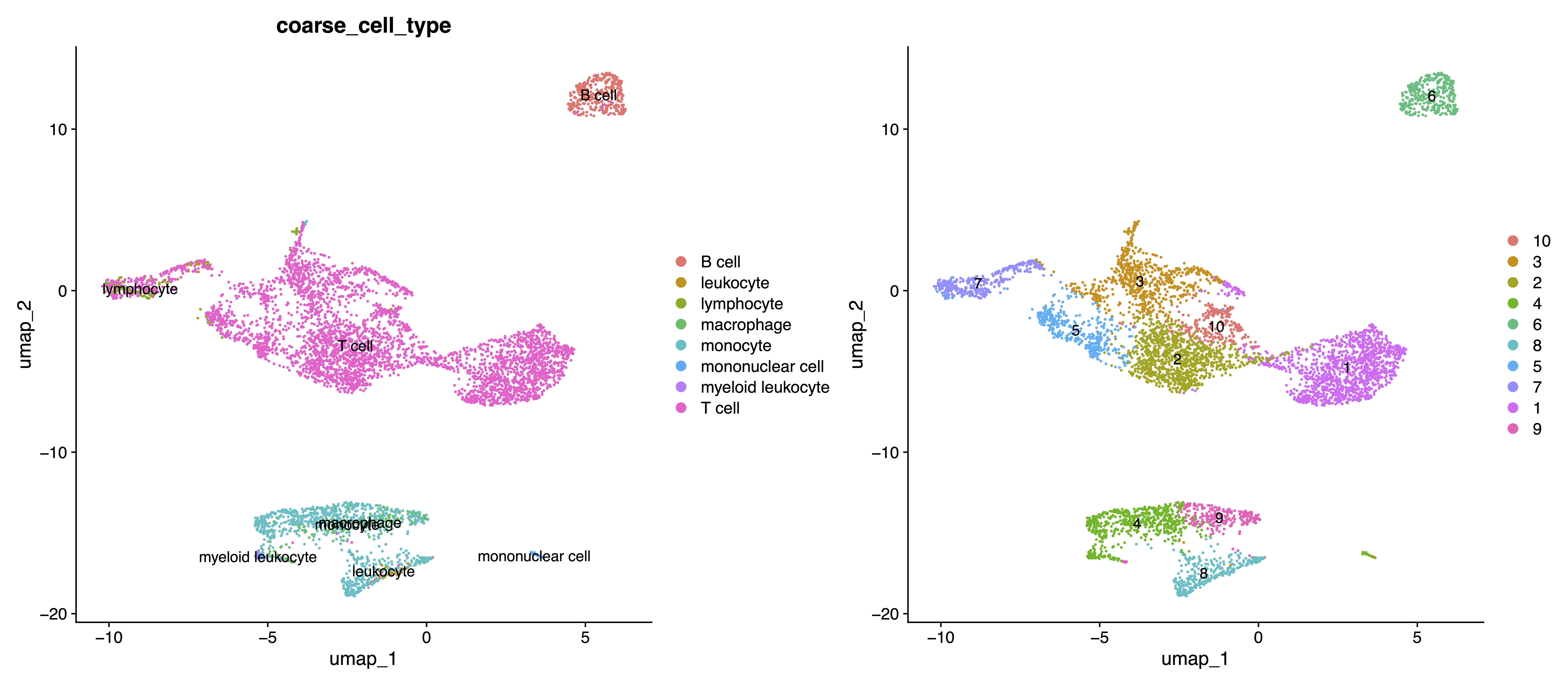
# Cell Ranger UMAP visualizations for cell types and clusters
plot3 <- DimPlot(seurat.hv10k, reduction="crumap", group.by = "coarse_cell_type", label=TRUE)
plot4 <- DimPlot(seurat.hv10k, reduction="crumap", label=TRUE)
pdf("./tests/pbmc5k/analysis/Cell_Ranger.dim_plot.cell_type.cluster.pdf", width = 16)
plot3+plot4
dev.off()
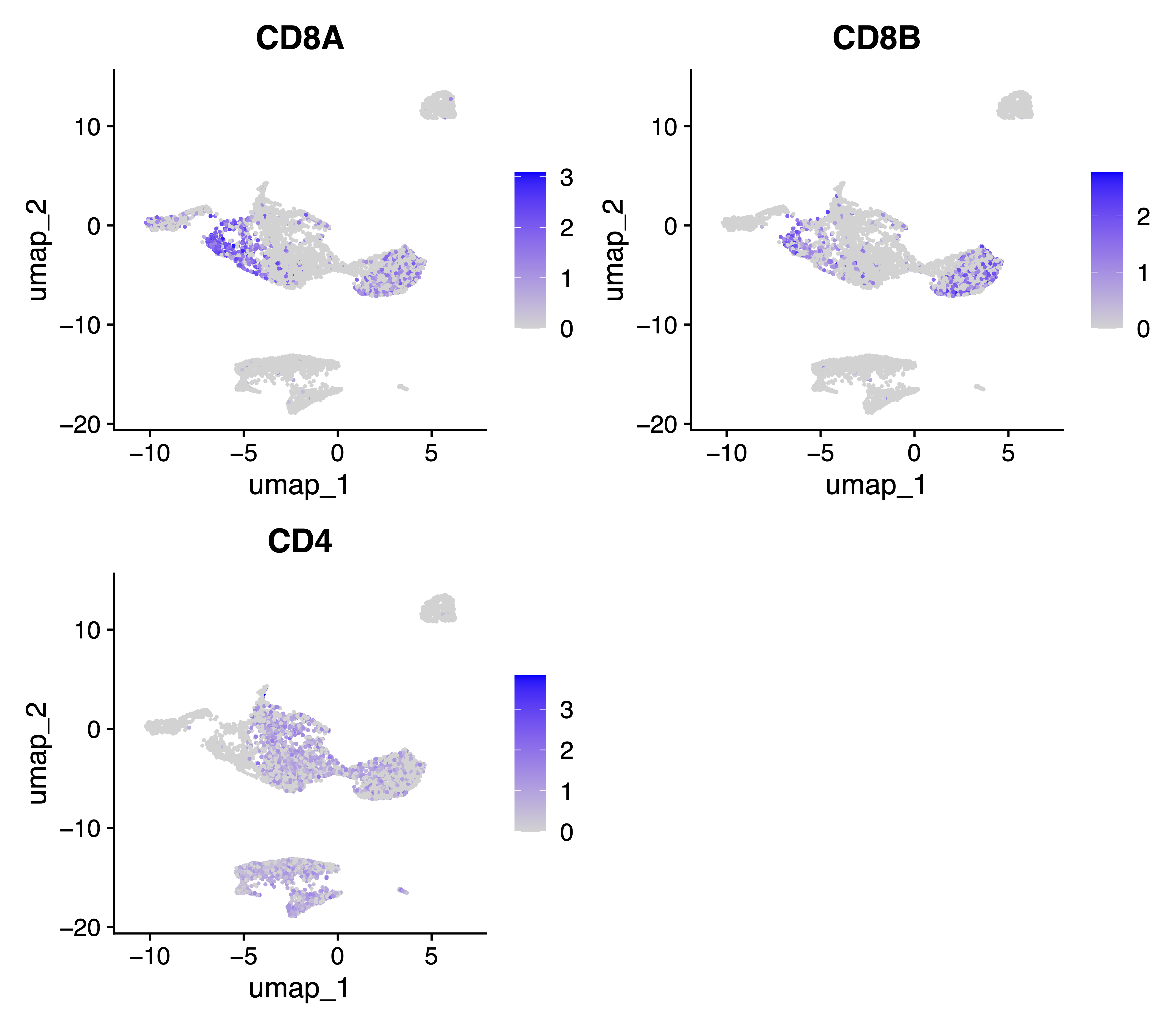
Also, we generate feature plots for key T cell marker genes CD8A, CD8B and CD4:
# feature plots for key T cell marker genes (CD8A, CD8B, CD4)
pdf("./tests/pbmc5k/analysis/Cell_Ranger.feature_plot.CD4.CD8.cluster.pdf", width = 8)
FeaturePlot(seurat.hv10k, features = c("CD8A", "CD8B", "CD4"))
dev.off()
Analysis SAKURA Embeddings with Seurat
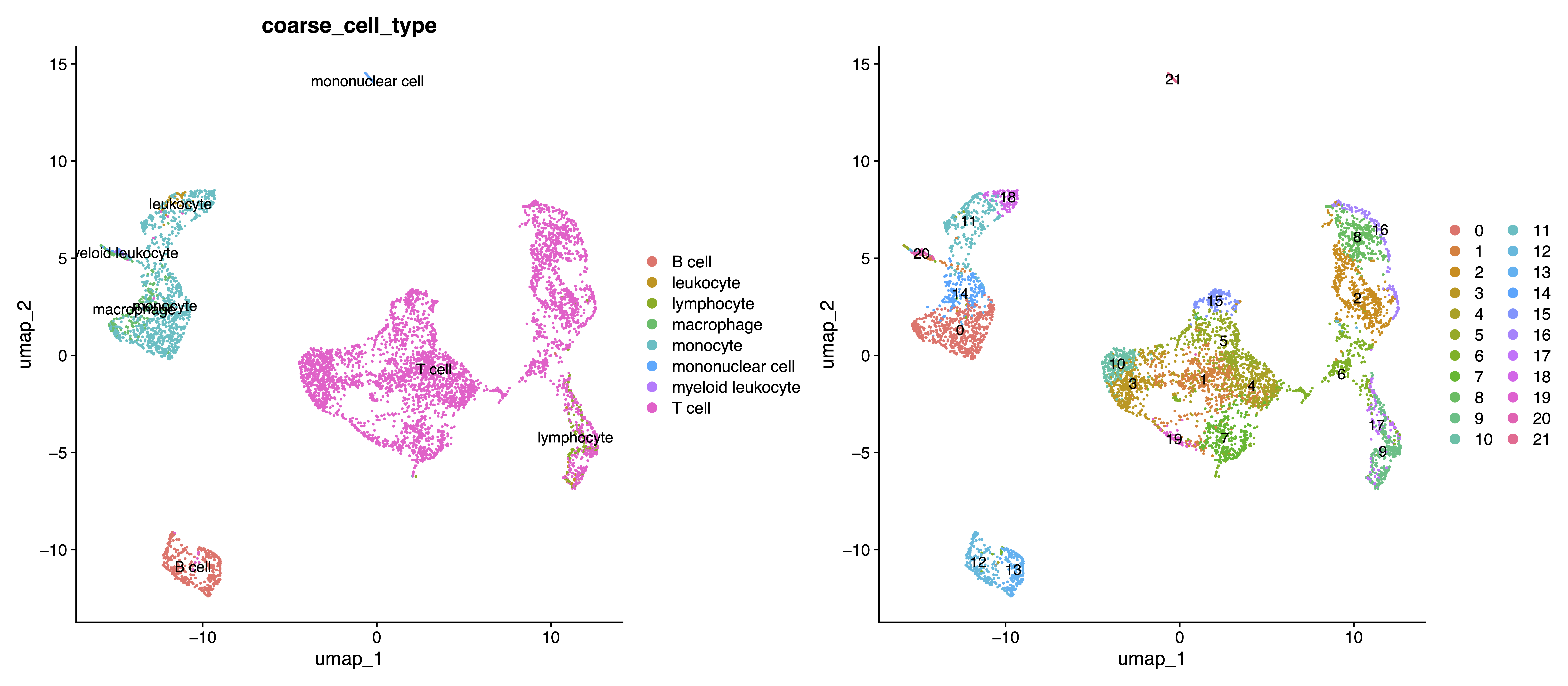
Then, we perform standard clustering and UMAP pipeline using Seurat on SAKURA cell embedding. Similarly, we generate comparative visualizations (UMAP) colored by Cell Ranger coarse cell type annotations and SAKURA cluster identities.
data <- read.csv('./test/pbmc5k/log/172_all_cell_all_latent.csv', row.names = 1)
embedding_data <- data[,1:50]
seurat.hv10k[["sakura"]] <- CreateDimReducObject(embeddings = embedding_data %>% as.matrix, key = "sakura_", assay = DefaultAssay(seurat.hv10k))
seurat.hv10k <- seurat.hv10k %>% FindNeighbors(reduction = 'sakura') %>% FindClusters()
seurat.hv10k <- RunUMAP(seurat.hv10k, reduction = "sakura", dims = 1:50)
dir.create("./tests/pbmc5k/analysis/")
plot1 <- DimPlot(seurat.hv10k, group.by = "coarse_cell_type", label=TRUE)
plot2 <- DimPlot(seurat.hv10k, label=TRUE)
# SAKURA UMAP visualizations for cell types and clusters
pdf("./tests/pbmc5k/analysis/SAKURA.dim_plot.cell_type.cluster.pdf", width = 16)
plot1+plot2
dev.off()
Also, we generate the same feature plots for key T cell marker genes CD8A, CD8B and CD4:
pdf("./tests/pbmc5k/analysis/SAKURA.feature_plot.CD4.CD8.cluster.pdf", width = 8)
FeaturePlot(seurat.hv10k, features = c("CD8A", "CD8B", "CD4"))
dev.off()
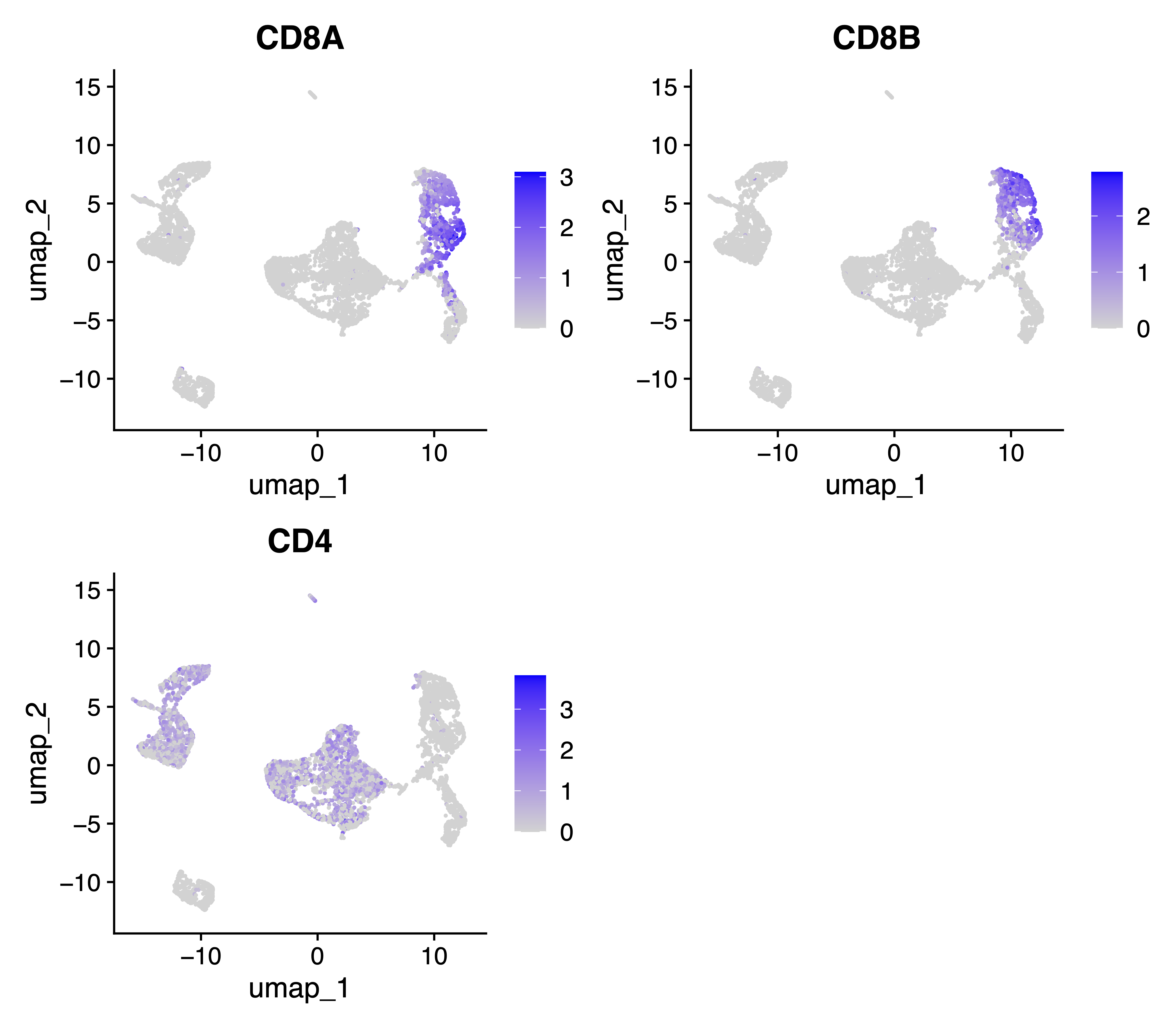
Comparing all the feature and UMAP plots from above analysis, the results indicate improved separation of CD4+ and CD8+ T cells without introducing stronger distortion to the SAKURA cell embedding, with the marker genes CD8A and CD8B as input knowledge.